![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
|
|
|
Services The pharmaceutical industry is heavily regulated and the MHRA in the UK (and the FDA in the US) licence drugs only if they meet stringent safety, quality and efficacy standards. These range from EU guides and annexes to Good Manufacturing Practice (GMP), Good Clinical Practice (GCP) and Good Laboratory Practice (GLP) to FDA codes of federal regulations, such as the 1997 "21 CFR Part 11 Electronic Records; Electronic Signatures" ruling. The MHRA and the FDA ensure drugs meet these standards through a process of regulation and enforcement. They have a number of powers to protect public health, ranging from working with the manufacturer of the product to legal remedies such as asking the manufacturer to recall a product, having agency officials seize products if a voluntary recall is not done and detaining imports at the port of entry until problems are corrected. If warranted, the FDA and MHRA can also ask the courts to issue injunctions or prosecute those that deliberately violate the law. Validation, therefore, has to be conducted to a standard that meets or exceeds those demanded by these agencies.
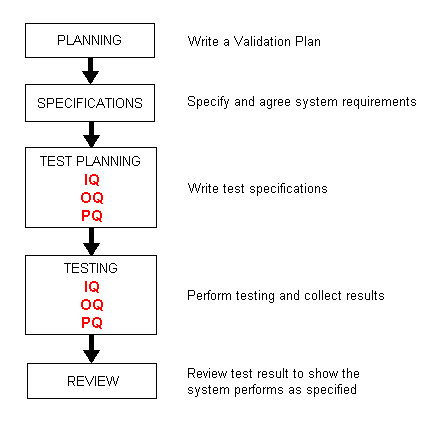
GAMP standard Validating computer systems to the GAMP standard satisfies this requirement. Established in 1991 and continually updated since in order to meet FDA expectations for compliance of manufacturing and related systems, GAMP defines the following validation lifecycle. The first stage is Planning.
This involves writing a Validation Plan (VP) to determine the validation
strategy to be adopted. A risk assessment of the system occurs during which
questions are asked such as: Does the system require validation? How much
validation is required? What aspects of the system are critical to product and
patient safety and to the business? The VP defines the validation activities,
the roles and responsibilities of the project team and the validation
deliverables. It is a key document and is approved by senior management. The second stage is Specifications. This involves creating documentation that describes system functionality. A User Requirements Specification (URS) describes what the system should do whilst a Functional Specification (FS) describes what the system will, or does, do. The third stage is Test Planning. This involves writing qualifications (test scripts) to verify, and challenge, the functionality of the system described in the specification documents. Traditionally, validation has comprised Installation Qualification (IQ), Operational Qualification (OQ) and Performance Qualification (PQ). The IQ provides verification that a system is installed according to approved specifications; the OQ provides verification that a system operates according to approved specifications; and the PQ provides verification that a system is capable of performing system processes according to approved specifications. The fourth stage is Testing. This involves running the test specifications (IQ, OQ, PQ) against the built system and documenting the results against predefined acceptance criteria described in the test specifications. Each test result should clearly state whether it has passed or failed. The tester and witness sign and date each test. The fifth stage is Review. This involves reviewing the test results and producing a Validation Report (VR). This summarises the validation activities, any deviations from the Validation Plan, outstanding and corrective actions and a statement of fitness for purpose of the system. Once a system is validated and in operation, plans and procedures should be defined to ensure the system remains in a validated state eg standard operating procedures, training and change control mechanisms.
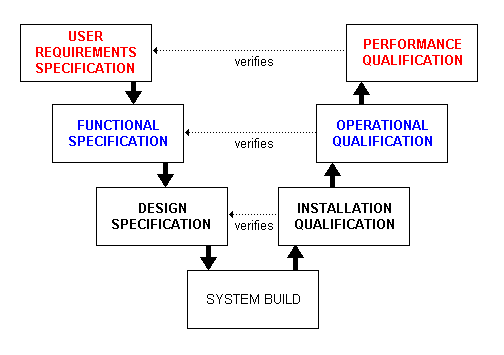
The GAMP standard defines the documents to be written and the activities to be undertaken to validate a system. It also places the validation lifecycle within the constraints of the classic V-model, shown to the left. For a validation project, this means that once the Validation Plan has been written (Planning), the URS may be created, followed by an FS and then a Hardware Design Specification (HDS) or Software Design Specification (SDS) or both, as is required (Specifications). Once these documents are approved (formally agreed), the system is built or delivered as appropriate. After this, the IQ, OQ and PQ tests are written (Test Planning) and executed (Testing) following approval. The test results are reviewed and if there are no deviations to investigate, the Validation Report is written and the system can go live (Review). It is then subject to formal change control in order to maintain its validated status.
The V-model is a powerful technique and offers many advantages over other formal models (eg waterfall) and informal methods:
How we can help We are fully experienced at writing all documents in the validation lifecycle to the GAMP standard (or company standards, if preferred). Whether it is reviewing existing documentation or writing all the documentation required for an entire project, we have the consultants with the necessary skills and expertise to do this on your behalf. We have written many examples of the technical specifications stipulated by the GAMP standard, including:
Similarly, we have produced all manner of test documentation:
We have validated many projects and can validate almost any computer system, whether it is a small bespoke package written in Visual Basic, a Microsoft Access database or a large multi-site ERP/MRP application. We do, however, boast particular knowledge and experience of validating the Documentum document management system, which we have been doing since 1996. Besides writing and reviewing validation documentation to GAMP standards, we also:
Any level of assistance can be provided, ranging from one-off single document creation to major projects requiring detailed and in-depth expertise. We also offer reviewing services to companies who have written validation documents in-house but require the reassurance of an independent expert. In fact, during the last decade we have helped several companies with their systems. NB Whilst our speciality is undoubtedly validation consultancy, our strength in document management, writing clear technical documents and testing validated systems has led to a number of non-validation projects. To see these engagements please see our Projects.
To see which companies we have assisted please refer to Clients. Or you may wish to return to the Home page by clicking on our company logo at the top of the page. |